In a recent post, Andrew Dalke compares SMILES parsing in Frowns (a now unsupported Open Source Python cheminformatics library) and the CDK (an actively developed Open Source Java cheminformatics library). Andrew is somewhat of a parsing expert - indeed, BioPython is built upon a parser called Martel which Andrew developed. The Frowns SMILES parser, contributed by Andrew, shows what an expert can do. It would be interesting to know whether similiar code could be incorporated into the CDK and OpenBabel, and if so, what is the speed tradeoff involved?
Andrew also discusses the compression of SMILES strings, which James Melville and Johnathan Hirst have also recently been studying. Rajarshi has also reviewed this work.
Friday, 29 June 2007
ANN: GaussSum 2.1.0 released

Announcing the release of GaussSum 2.1.0.
GaussSum is a GUI application that can analyse the output of ADF, GAMESS (US), GAMESS-UK, Gaussian, Jaguar and PC GAMESS to extract and calculate useful information. This includes the progress of the SCF cycles, geometry optimisation, UV-Vis/IR/Raman spectra, MO levels, MO contributions and more. [Full description]
Release Notes for Version 2.1.0:
* Major update in parsing library (upgraded from cclib 0.6.1 to
cclib 0.8dev)
* Jaguar log files now supported
* Compressed log files (.zip, .gz., .bz2) supported
* Underlying code now uses Numpy for numerical calculation, rather
than the deprecated Numeric (see install notes)
* Fixed error in the output of DOS_spectrum.txt where the 'Total'
column was equal to the values for the first group
* Fixed problem plotting the COOP
* Fixed plotting the UV-Vis spectrum, if there were no groups
* Corrected the documentation
Thanks to everyone who reported bugs.
Friday, 22 June 2007
4th Joint Sheffield Conference on Chemoinformatics
I recently attended the 4th Joint Sheffield Conference on Chemoinformatics, and enjoyed it a lot.
Some of the things I noted were:
(1) Industry talks have suddenly got more interesting (than they were, that is). After describing methods run on their own in-house data, they suddenly say "In order to compare with other methods, I ran this method on a publicly available dataset". Great. About time. The big free datasets are now so well established, they've even heard about them in industry. Thanks go to ZINC, DUD, PubChem, and the NIH guys who are even making available some HTS data (is this correct? I cannot easily find it on the web).
(2) In this postmodern age, it is now a requirement for all cheminformatics conferences to start with a talk that tells us we're wasting our time trying to dock anything, as it doesn't work. Full marks for shock value, but perhaps the more interesting content of Anthony Nicholls talk related to statistical comparisons of AUC (area under the ROC curve) for a published study of multiple docking problems (Warren et al.). Basically, the error bars are so large we cannot say that any program is significantly different from any other (according to him :-) [disclaimer, I'm developing GOLD]).
(3) Of course, no cheminformatics conference would be complete without dodgy statistics, and I'm as guilty of it (not knowingly, I hope) as anyone. Multiple tests on the same dataset require corrections for significance testing such as the Bonferroni statistic - "if it passes the Bonferroni it's probably true" was the quote from Martin Packer (AZ). Everyone wrote that one down when it was mentioned. But for the cheminformaticians who skipped Statistics 101, there was more extra homework. Jonathan Hirst directed us to read the appendix of one of his papers for some more light reading on hard-core statistics such as the Nemenyi test and the improved Friedman statistic.
(4) Open Source chemistry software got a mention by some of the academics speaking. Jonathan Hirst in particular gave Joelib2 a big thumbs up, and made it clear that his own software is Openly available from his web page (although no license is mentioned in the README there). The author of Joelib2 was in the audience, Joerg Kurt Wegner, and it would have been nice if the speaker had put Joerg's name on his slide along with the name of the program and the website. After all, it's nice to get some personal recognition if you put a lot of work into such a program and then make it Openly available. Jonathan had to skip his next-to-last slide promoting the Blue Obelisk group, but it was still good to see the reference flash by. Irilenia Nobeli used the CDK, as did David Wild who is very active in the development of Web services with open source software.
Some of the things I noted were:
(1) Industry talks have suddenly got more interesting (than they were, that is). After describing methods run on their own in-house data, they suddenly say "In order to compare with other methods, I ran this method on a publicly available dataset". Great. About time. The big free datasets are now so well established, they've even heard about them in industry. Thanks go to ZINC, DUD, PubChem, and the NIH guys who are even making available some HTS data (is this correct? I cannot easily find it on the web).
(2) In this postmodern age, it is now a requirement for all cheminformatics conferences to start with a talk that tells us we're wasting our time trying to dock anything, as it doesn't work. Full marks for shock value, but perhaps the more interesting content of Anthony Nicholls talk related to statistical comparisons of AUC (area under the ROC curve) for a published study of multiple docking problems (Warren et al.). Basically, the error bars are so large we cannot say that any program is significantly different from any other (according to him :-) [disclaimer, I'm developing GOLD]).
(3) Of course, no cheminformatics conference would be complete without dodgy statistics, and I'm as guilty of it (not knowingly, I hope) as anyone. Multiple tests on the same dataset require corrections for significance testing such as the Bonferroni statistic - "if it passes the Bonferroni it's probably true" was the quote from Martin Packer (AZ). Everyone wrote that one down when it was mentioned. But for the cheminformaticians who skipped Statistics 101, there was more extra homework. Jonathan Hirst directed us to read the appendix of one of his papers for some more light reading on hard-core statistics such as the Nemenyi test and the improved Friedman statistic.
(4) Open Source chemistry software got a mention by some of the academics speaking. Jonathan Hirst in particular gave Joelib2 a big thumbs up, and made it clear that his own software is Openly available from his web page (although no license is mentioned in the README there). The author of Joelib2 was in the audience, Joerg Kurt Wegner, and it would have been nice if the speaker had put Joerg's name on his slide along with the name of the program and the website. After all, it's nice to get some personal recognition if you put a lot of work into such a program and then make it Openly available. Jonathan had to skip his next-to-last slide promoting the Blue Obelisk group, but it was still good to see the reference flash by. Irilenia Nobeli used the CDK, as did David Wild who is very active in the development of Web services with open source software.
Sunday, 17 June 2007
Use Linux on Windows (for free!)
My Dell laptop runs WinXP, but I find it useful to have access to Linux. Here are the steps you need to take to run a Linux distribution from Windows using VMWare, who provide VMWare Player (and also VMWare Server) for free.
I find this method of accessing Linux useful:
(1) because I need to develop software on Linux and Windows
(2) it's nicer than Putty if you need to 'ssh' somewhere
(3) you don't need to reboot your computer to use Linux
(4) you can close the virtual machine and it starts up at the same point next time
(5) if you use VMWare Server, you can take snapshots of your entire operating system and state, which you can continue from later or give to other people
(6) no need to install Linux yourself or repartition your hard drive
(7) security -- let the hackers hack your virtual machine rather than your desktop PC
(8) you can test webservices, as the virtual machine behaves like a completely different PC
It's pretty easy to setup:
(1) Download and install VMWare Player from the link above
(2) Download a virtual machine containing a Linux distribution from the VMWare site (for example). I downloaded a virtual machine of Debian Etch. What's good about Debian is that it's stable, and has a really nice package installation system containing 19000 packages.
(3) Start the virtual machine by opening in VMWare Player, and sit back while it boots up.
(4) The one difficulty with VMWare is getting the networking to work. As described on the home page for this virtual machine, you need to start the VMWare network driver by entering the following as root (the passwords for 'user' and 'root' and simply 'user' and 'root'):
If it worked, the output of '/sbin/ifconfig' should show eth0 as a network driver, and it should be possible to access the internet, and 'ssh' out of the machine.
(5) At this point, you can start installing whatever packages you want, by clicking on "Desktop", "Administration", "Synaptic Package Manager" (or use the command-line interface, 'apt-get install whatever')
Here's the obligatory screenshot showing me writing this blog post on Windows, and running VMWare/Debian at the same time. I've just installed and run GaussSum at the command line:

I find this method of accessing Linux useful:
(1) because I need to develop software on Linux and Windows
(2) it's nicer than Putty if you need to 'ssh' somewhere
(3) you don't need to reboot your computer to use Linux
(4) you can close the virtual machine and it starts up at the same point next time
(5) if you use VMWare Server, you can take snapshots of your entire operating system and state, which you can continue from later or give to other people
(6) no need to install Linux yourself or repartition your hard drive
(7) security -- let the hackers hack your virtual machine rather than your desktop PC
(8) you can test webservices, as the virtual machine behaves like a completely different PC
It's pretty easy to setup:
(1) Download and install VMWare Player from the link above
(2) Download a virtual machine containing a Linux distribution from the VMWare site (for example). I downloaded a virtual machine of Debian Etch. What's good about Debian is that it's stable, and has a really nice package installation system containing 19000 packages.
(3) Start the virtual machine by opening in VMWare Player, and sit back while it boots up.
(4) The one difficulty with VMWare is getting the networking to work. As described on the home page for this virtual machine, you need to start the VMWare network driver by entering the following as root (the passwords for 'user' and 'root' and simply 'user' and 'root'):
/etc/init.d/networking stop
rmmod pcnet32
rmmod vmxnet
depmod -a
modprobe vmxnet
/etc/init.d/networking start
If it worked, the output of '/sbin/ifconfig' should show eth0 as a network driver, and it should be possible to access the internet, and 'ssh' out of the machine.
(5) At this point, you can start installing whatever packages you want, by clicking on "Desktop", "Administration", "Synaptic Package Manager" (or use the command-line interface, 'apt-get install whatever')
Here's the obligatory screenshot showing me writing this blog post on Windows, and running VMWare/Debian at the same time. I've just installed and run GaussSum at the command line:

Friday, 15 June 2007
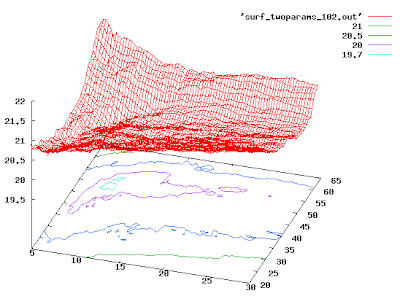
Plot data in 3D
You can use Gnuplot to draw surfaces and contours. You need to get your data into the following grid format first:
Once you've done this, here are the magic Gnuplot commands:
At this point, you should click on the graph and spin it around until it looks nice. Then you can save it to disk as a PNG (for better quality, use PS output):
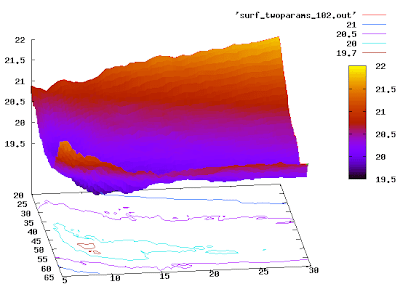
If you want it even nicer, you can try:
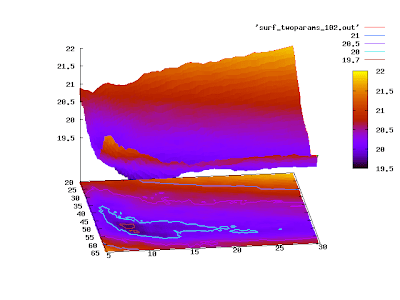
or
x1, y1, val1
x1, y2, val2
...
x1, yN, valN
[blank line]
x2, y1, val(N+1)
x2, y2, val(N+2)
...
x2, yN, val(2N)
[blank line]
...and so on...
Once you've done this, here are the magic Gnuplot commands:
splot 'mygriddata.txt' with lines
set contour
set cntrparam levels discrete 19.7, 20, 20.5, 21, 22, 23, 24
replot
At this point, you should click on the graph and spin it around until it looks nice. Then you can save it to disk as a PNG (for better quality, use PS output):
set terminal png
set output 'plot.png'
replot

If you want it even nicer, you can try:
set pm3d
set hidden3d
replot

or
set pm3d at sb
replot

Thursday, 14 June 2007
See you in Sheffield?
Conference season starts Monday for me. I'll be attending the Sheffield Conference on Cheminformatics on 18th June, and if you are too, be sure to say hi.
Friday, 8 June 2007
Installing Mediawiki on Sourceforge
Installing Mediawiki on Sourceforge requires some extra effort compared to a normal Mediawiki installation, as Sourceforge restricts certain activites on the web server and you need to work around these. In case you ever need to do this, here is a summary of what I did when I installed Mediawiki for BlueObelisk.org.
Follow László Monda's instructions with the following changes:
Follow László Monda's instructions with the following changes:
- In Step 1, you should only download a version of Mediawiki that will run with PHP4 (since PHP5 is not available on Sourceforge). I used Mediawiki-1.6.7.
- Forget the line numbers in Step 3, just search for the relevant line. The text described as being on line 1125 did not exist in the file. Instead, you should change the line (line 383 in my installation):
$conf->IP = dirname( dirname( __FILE__ ) );
to$conf->IP = "/home/groups/p/pr/projectname/htdocs/wiki";
- In Step 5, I used 'true' for $wgGroupPermissions['user']['edit']. You should place the logo in htdocs, and edit $wgLogo to point to "/mylogo.png", or whatever.
Minimizing an objective function using Python
Thanks largely to physicists, Python has very good support for efficient scientific computing. The following code shows how to use the brute-force optimization function of scipy to minimize the value of some objective function with 4 parameters. Since it is a grid-based method, it's likely that you may have to rerun the optimization with a smaller parameter space.
A number of cleverer optimization functions are also available in scipy, including constrained optimization and stochastic methods. For more information, install scipy (on Windows or Linux), and type the following at the Python prompt:
import numpy
import scipy.optimize
def my_objective_fn(params):
print params,
value_of_objectivefn = calc_objectivefn_value(params)
print value_of_objectivefn
return value_of_objectivefn
if __name__=="__main__":
# Method 1 (same number of steps for each parameter)
myranges = ((21,49), (0,0.1), (95,123), (1.6, 2.0))
scipy.optimize.brute(my_objective_fn, myranges, Ns=5)
# Method 2 (can have different number of steps for
# different parameters)
slice_obj = numpy.s_[21:56:7, 0:0.1:0.02,
95:130:7, 1.6:2.1:0.1]
scipy.optimize.brute(my_objective_fn, slice_obj)
A number of cleverer optimization functions are also available in scipy, including constrained optimization and stochastic methods. For more information, install scipy (on Windows or Linux), and type the following at the Python prompt:
>>> import scipy.optimize
>>> help(scipy.optimize)
DESCRIPTION
Optimization Tools
==================
A collection of general-purpose optimization routines.
fmin -- Nelder-Mead Simplex algorithm
(uses only function calls)
fmin_powell -- Powell's (modified) level set method
(uses only function calls)
...
Monday, 4 June 2007
Add to Connotea from Journal Pages
If you regularly read journal Table of Contents pages and want an easy way to keep track of papers you find interesting, you may want to try my "Add to Connotea" Greasemonkey script.

To use it, you first need to have a Connotea account. When you navigate to a journal web page for the first time after you have started your browser, you will need to enter your Connotea username and password.
The script adds an "Add to Connotea" link next to every DOI on journal pages. If you click on this, a dialog box appears that allows you to bookmark the paper on Connotea. A useful feature is the ability to quickly select from tags that you have already used. The "Add to Connotea" script also displays a count of the number of people who have already bookmarked a particular paper. If you click on this number, it will bring you to the Connotea page for that paper.
Why use Connotea inside of just bookmarking a paper, or saving it on your computer? Here's why I do: adding to Connotea is 'cheap'. I'm a lazy bookmarker; now that my bookmarks have reached the bottom of the my screen I've given up. Arranging into folders is too much hassle for my busy lifestyle. Now I can tag a paper with "Read me later", or "Journal club", and I will actually be able to find them later. Bookmarks are available from any browser; you can bookmark from home, and read the paper later from work. Your computer may explode (not due to my Greasemonkey script, I hope), but your bookmarks will still exist. Another nice thing is that Connotea is part of Web 2.0 - it provides an API that allows it to be used in mashups; like this Greasemonkey script!
There is also the 'social' side to 'social bookmarking'. I have already mentioned that the "Add to Connotea" script keeps a count of how many people have bookmarked a particular article; this may indicate that a particular article is worth reading. There are people who track all articles posted with particular tags, e.g. "evolution", to keep up to date with the latest articles, but I'm not sure how useful this is in chemistry. Also, you may want to keep up to date with what all the kool kids are reading, in which case you may want to track what articles your friends are bookmarking (note: all bookmarks are generally public, but you can opt to make them private). Connotea allows you to add a description and/or a comment to an article, but while this seems like a neat idea (e.g. for posting a reviews), it does not seem to be used that much and in fact, seems to suffer from link spam (see here).
I should point out that Connotea also provides a bookmarklet that you can click after selecting a DOI. While the bookmarklet is the only option for websites that my Greasemonkey script can't handle, I find my script a handier way to bookmark papers on journal websites, plus you have the added goodness of identifying popular articles.

To use it, you first need to have a Connotea account. When you navigate to a journal web page for the first time after you have started your browser, you will need to enter your Connotea username and password.
The script adds an "Add to Connotea" link next to every DOI on journal pages. If you click on this, a dialog box appears that allows you to bookmark the paper on Connotea. A useful feature is the ability to quickly select from tags that you have already used. The "Add to Connotea" script also displays a count of the number of people who have already bookmarked a particular paper. If you click on this number, it will bring you to the Connotea page for that paper.
Why use Connotea inside of just bookmarking a paper, or saving it on your computer? Here's why I do: adding to Connotea is 'cheap'. I'm a lazy bookmarker; now that my bookmarks have reached the bottom of the my screen I've given up. Arranging into folders is too much hassle for my busy lifestyle. Now I can tag a paper with "Read me later", or "Journal club", and I will actually be able to find them later. Bookmarks are available from any browser; you can bookmark from home, and read the paper later from work. Your computer may explode (not due to my Greasemonkey script, I hope), but your bookmarks will still exist. Another nice thing is that Connotea is part of Web 2.0 - it provides an API that allows it to be used in mashups; like this Greasemonkey script!
There is also the 'social' side to 'social bookmarking'. I have already mentioned that the "Add to Connotea" script keeps a count of how many people have bookmarked a particular article; this may indicate that a particular article is worth reading. There are people who track all articles posted with particular tags, e.g. "evolution", to keep up to date with the latest articles, but I'm not sure how useful this is in chemistry. Also, you may want to keep up to date with what all the kool kids are reading, in which case you may want to track what articles your friends are bookmarking (note: all bookmarks are generally public, but you can opt to make them private). Connotea allows you to add a description and/or a comment to an article, but while this seems like a neat idea (e.g. for posting a reviews), it does not seem to be used that much and in fact, seems to suffer from link spam (see here).
I should point out that Connotea also provides a bookmarklet that you can click after selecting a DOI. While the bookmarklet is the only option for websites that my Greasemonkey script can't handle, I find my script a handier way to bookmark papers on journal websites, plus you have the added goodness of identifying popular articles.
Saturday, 2 June 2007
Open Babel Python module for Windows (1.2) released
Announcing the release of the OpenBabel module for Python, version 1.2, for Windows.
OpenBabel is a chemical toolbox designed to speak the many languages of chemical data. It's an open, collaborative project allowing anyone to search, convert, analyze, or store data from molecular modeling, chemistry, solid-state materials, biochemistry, or related areas. This Python module allows you to access this popular C++ library in your Python scripts.
New features:
(1) Data fields in file formats like MOL2 and SDF can now be accessed and edited
(2) Unit cell data in crystallographic file formats such as CIF can now be accessed
(3) A list of all detected input and output file formats can be accessed
Core features:
(1) Read and write over 80 molecular file formats
(2) Access molecular properties like molecular weight, formula, charge
(3) Daylight-type fingerprints and calculation of the Tanimoto coefficient
(4) SMARTS pattern matching
(5) Graph algorithms such as the Smallest Set of Smallest Rings (SSSR) and Depth-First Iteration over atoms
Documentation:
(1) The Python module
(2) The C++ toolkit
(3) The OpenBabel web site
Support:
(1) If you have any questions, send an email to openbabel-scripting@lists.sourceforge.net
(2) Report a bug
P.S. The Python module is also available for Linux and MacOSX. It is also possible to use OpenBabel from C++, Perl, Ruby and Java, although getting these to work on Windows may be more difficult.
OpenBabel is a chemical toolbox designed to speak the many languages of chemical data. It's an open, collaborative project allowing anyone to search, convert, analyze, or store data from molecular modeling, chemistry, solid-state materials, biochemistry, or related areas. This Python module allows you to access this popular C++ library in your Python scripts.
New features:
(1) Data fields in file formats like MOL2 and SDF can now be accessed and edited
(2) Unit cell data in crystallographic file formats such as CIF can now be accessed
(3) A list of all detected input and output file formats can be accessed
Core features:
(1) Read and write over 80 molecular file formats
(2) Access molecular properties like molecular weight, formula, charge
(3) Daylight-type fingerprints and calculation of the Tanimoto coefficient
(4) SMARTS pattern matching
(5) Graph algorithms such as the Smallest Set of Smallest Rings (SSSR) and Depth-First Iteration over atoms
Documentation:
(1) The Python module
(2) The C++ toolkit
(3) The OpenBabel web site
Support:
(1) If you have any questions, send an email to openbabel-scripting@lists.sourceforge.net
(2) Report a bug
P.S. The Python module is also available for Linux and MacOSX. It is also possible to use OpenBabel from C++, Perl, Ruby and Java, although getting these to work on Windows may be more difficult.
Subscribe to:
Posts (Atom)