First of all, here's the before picture:

pybel.Molecule._repr_png_ = lambda x: x.write("png")And here's how it looks after:
pybel.Molecule._repr_png_ = lambda x: x.write("png")And here's how it looks after:
classpath.append("/C:/workspace/Jython/lib/cdk-1.4.5.jar");If there's someone from Knime reading this, please let me know the correct way of doing this. I know that there's an extension point for extending the Classpath of the Jython node, but how is this used?

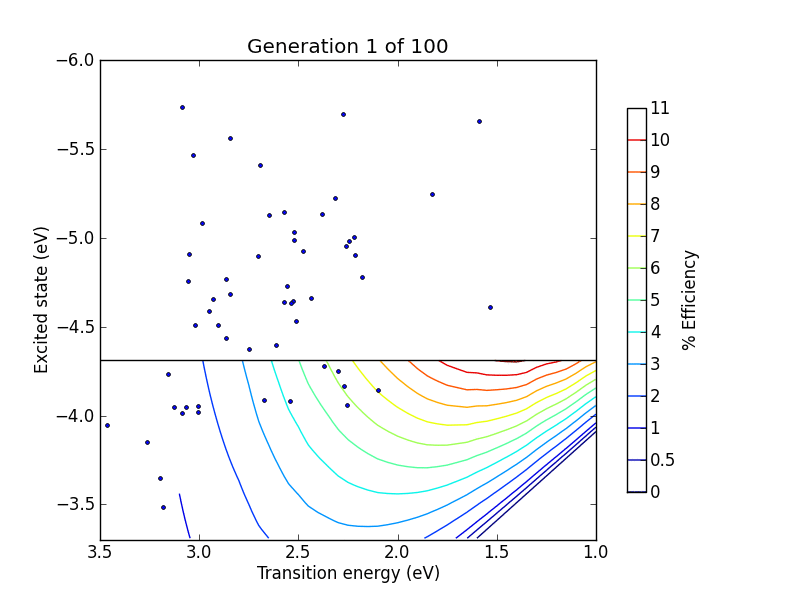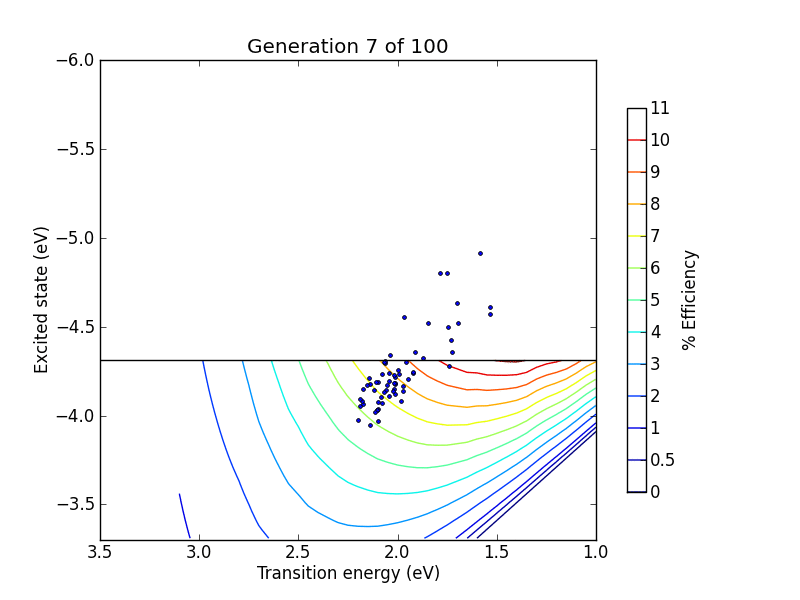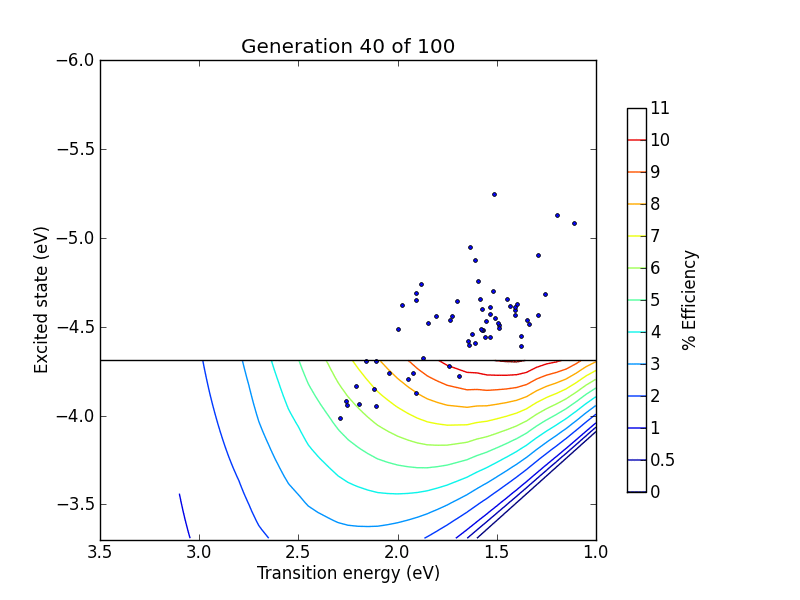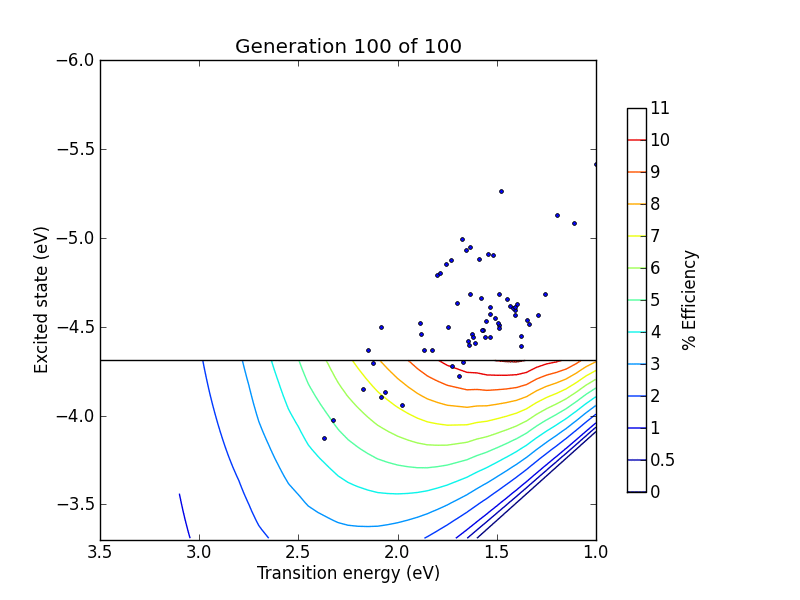
vals = range(0, 10) + range(19, 100 ,10)
for N in vals:
homo, trans = self.xy[N]
lumo = [x+y for x,y in zip(homo,trans)]
pylab.title("Generation %d of 100" % (N+1,))
pylab.plot(trans, lumo, ".")
pylab.savefig("pictures/animation%02d.png" % N)
pylab.clf()
You can then use ImageMagick to do the PNG to animated GIF conversion. ImageMagick is a bit like Open Babel - it is a one-stop shop for file format conversion, plus it can do a whole set of transformations, and furthermore can be accessed as a programming library. It is available cross-platform (I used it on Windows). The main binary is called "convert", and the conversion is done as follows:convert.exe -delay 20 -loop 0 animation*.png GA_animation.gifYou can also specify different timings for every frame as I did above. I wrote a little script to help with this:
export OB=$HOME/openbabel sudo apt-get update sudo apt-get install cmake subversion g++ swig2.0 python-dev libxml2-dev wx-common wx2.8-headers libwxbase2.8-dev libeigen3-dev libwxgtk2.8-dev mkdir $OB cd $OB svn checkout http://openbabel.svn.sourceforge.net/svnroot/openbabel/openbabel/trunk trunk mkdir build cd build cmake ../trunk -DCMAKE_INSTALL_PREFIX=../install -DPYTHON_BINDINGS=ON -DRUN_SWIG=ON make -j2 && make install export PATH=$OB/install/bin:$PATH export PYTHONPATH=$OB/install/lib:$PYTHONPATHThereafter, just type "svn update" in $OB/trunk, and "make && make install" in $OB/build, to get the latest and greatest.
Group contribution descriptors are a common type of molecular descriptor whose value is a sum of contributions from substructures of the molecule. Such a descriptor can easily be added to Open Babel without the need to recompile the code. All you need is a set of SMARTS strings for each group, and their corresponding contributions to the descriptor value.To read the rest, check out the development docs.
The following example shows how to add a new descriptor, hellohalo, whose value increments by 1, 2, 3 or 4 for each F, Cl, Br, and I (respectively) in the molecule...