In the
previous article, I showed how to install Jmol. This article will look at depicting a ligand in a protein active site with a few extra complications. This example was not chosen to showcase Jmol - this is a real-life example of a figure I need to create. You can find on the web lots of examples of scripting Jmol to turn on/off ball-and-stick models. Figuring out how to do difficult things is a bit harder, and I hope others will find this example useful...
Specifically, I want to depict the ligand in the protein active site, with the protein represented by an isosurface coloured by depth in the protein (values which I will supply), and with protein-ligand hydrogen bonds shown. I have a protein
.MOL2 file with hydrogens added; I have a ligand
.MOL file, I have the corresponding protein
.PDB file (no hydrogens and the ligand and waters have been removed); and I have
a text file containing, for each atom in the protein .MOL2 file, a value related to the depth of that atom in the protein. And here is the final image (screenshotted from the browser , cropped a little, and saved as a JPG with zero compression - click for the original image):
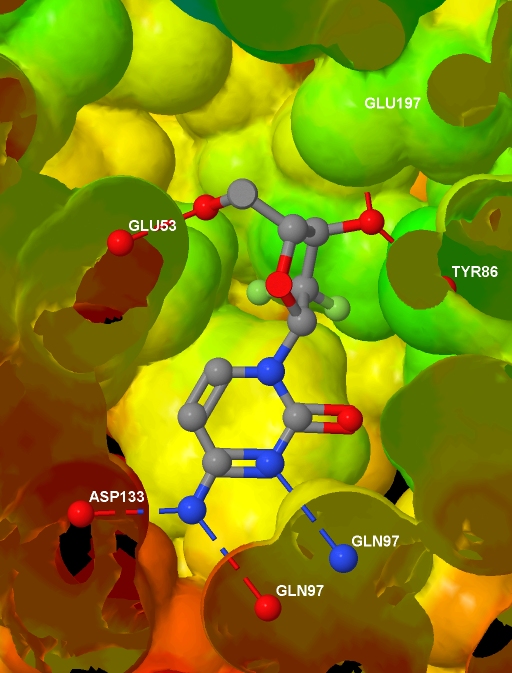
Before presenting the necessary code, to help understanding I note that Jmol imposes some constraints:
- bonds can only exist between molecules in the same model
- if the slab is turned on, it cuts through every model
- Jmol can identify the residue names in the PDB file, but not in the .MOL2 file
- Jmol does not automatically identify protein-ligand hydrogen bonds
- if mapping property data onto an isosurface, it needs to be supplied for every atom in the model (actually, there is an alternative, but this is a good rule of thumb)
I should also point out that Jmol can colour isosurfaces by depth in the protein automatically, but I wanted to use my own measure of depth.
Update 13/12/07: Due to popular demand, here is the actual applet in motion: [
low quality] [
high quality]. And finally, after all that, here is the code (available also as a
download):
<html>
<head>
<script type="text/javascript"
src="file:///D:/Tools/Jmol/jmol-11.3.53/Jmol.js">
</script>
</head>
<body>
<script type="text/javascript">
var protein = "1p62";
jmolInitialize("file:///D:/Tools/Jmol/jmol-11.3.53", true);
var jmolcmds = [
// Load the molecules
"load file:///D:/Ligands/" + protein + "_ligand.mol",
"set appendNew = false",
"load APPEND file:///D:/Proteins/" + protein + "_protein.mol2",
"set appendNew = true",
"load APPEND file:///D:/Proteins/" + protein + ".pdb",
// Turn up display quality but don't yet show anything
"display none; set antialiasDisplay true",
// The following is the output of "show orientation"
"reset;center {67.99645 35.5767 19.92025}",
"rotate z -21.47; rotate y 25.81; rotate z 74.65",
"zoom 400.0; set rotationRadius 35.03",
// Add the HBonds
"model 1.1",
"connect 3.1 (molecule=1 and elemno=8) \
(molecule!=1 and elemno=7) HBOND RADIUS 0.07 CREATE",
"connect 3.2 (molecule=1 and elemno=7) \
(molecule!=1 and elemno=8) HBOND RADIUS 0.07 CREATE",
"connect 3.1 (molecule=1 and elemno=8) \
(molecule!=1 and elemno=8) HBOND RADIUS 0.07 CREATE",
"connect 3.1 (molecule=1 and elemno=7) \
(molecule!=1 and elemno=7) HBOND RADIUS 0.07 CREATE",
// Load info for isosurface and draw it
"{model=1.1}.property_d = load(\"D:/Data/" + protein +"_rd.txt\")",
"isosurface select(molecule!=1 and within(10.0, molecule=1)) \
ignore(molecule=1) resolution 5 sasurface 0 \
COLOR ABSOLUTE 30 120 REVERSECOLOR MAP property_d",
// Select and label the HBonded protein atoms in model 2.1
"define hbondedproteinatoms within(3.2, true, \
model=1.1 and molecule=1 and (elemno=7 or elemno=8)) \
and not within(0.1, true, model=1.1 and molecule=1) \
and model=2.1 and not elemno=1 and not solvent",
"select hbondedproteinatoms; font label 15 SANSERIF BOLD",
"set labelFront; color label white; label %n%R",
// Add to selection the ligand and
// its HBonded protein atoms in model 1.1
"select selected or (((molecule=1 and model=1.1) or \
within(3.2, molecule=1 and model=1.1 and \
(elemno=7 or elemno=8))) and not elemno=1)",
// Slice through everything with a slab
"slab on; slab 51",
// Remove the Jmol logo (I will reference Jmol instead)
"set frank off",
// Show the selected data from all models
"model 0; display selected",
];
jmolApplet(700, jmolcmds.join("; "));
</script>
</body>
</html>


